
Obstructive Sleep Apnea and Cardiovascular Disease: Back and Forward in Time Over the Last 25 Years
Stuart F. Quan, M.D.
Division of Sleep Medicine, Harvard Medical School and Brigham Womens Hospital, Boston, MA; Arizona Respiratory Center, University of Arizona College of Medicine, Tucson, AZ
Abstract
Over the past 25 years, there have been significant advances made in understanding the pathophysiology and cardiovascular consequences of obstructive sleep apnea (OSA). Substantial evidence now implicates OSA as an independent risk factor for the development of hypertension, coronary artery disease, congestive heart failure and stroke, as well as increased risk of death. Pathophysiologic mechanisms include release of inflammatory mediators, oxidative stress, metabolic dysfunction, hypercoagulability and endothelial dysfunction. Although non-randomized intervention studies suggest that treatment of OSA with continuous positive airway pressure may mitigate its impact of the development of cardiovascular disease, randomized clinical trials are lacking.
Introduction
The first report of the physiologic events occurring in obstructive sleep apnea (OSA) was published in 1965 by Gastaut and colleagues (1). However, literary and historical accounts of what most likely was OSA have existed since antiquity (2). Most well-known of these is the description of Burwell’s case of a man falling asleep while playing poker. This led to the widespread use of the “Pickwickian Syndrome” because of the similarity of the patient’s symptoms to the literary imagery of “Joe, the fat boy”, a character in Charles Dicken’s Posthumous Papers of the Pickwick Club (3). In the 50 years since Gastaut’s original description of OSA, there has been an exponential growth in the recognition that it is a clinical condition with a high prevalence, subtle to disabling clinical symptoms, and more recently, substantial cardiovascular morbidity and mortality. However, much of the progress in our understanding of OSA with respect to cardiovascular disease has occurred in the past 25 years. Thus, it is appropriate to look back to where we were 25 years ago, our current state of knowledge and identify avenues for future research. Before doing so, however, one must examine whether there is sufficient evidence suggest that a biologic association between OSA and cardiovascular disease is plausible.
Obstructive Sleep Apnea and Cardiovascular Disease: Biologic Plausibility
Obstructive sleep apnea is characterized by repetitive episodes of occlusion or near occlusion of the upper airway at the level of the pharynx despite increasing inspiratory efforts (4). These episodes are terminated by brief arousals from sleep resulting in sleep fragmentation. There are a number of physiologic consequences to what essentially are repetitive involuntary Müeller maneuvers (5,6). With cessation or near cessation of airflow, transient oxygen desaturation and hypercarbia occur (4,6). Inspiratory efforts against an occluded airway result in large intrathoracic pressure swings (5,6). As a result, there is increased sympathetic nervous system activity (7), fluctuations in parasympathetic tone (8), large cyclical changes in heart rate, arterial and pulmonary vasoconstriction and hypertension, and increases in cardiac preload and afterload (9-11). Given the chronicity of OSA, it is not difficulty project that such physiologic changes might lead to daytime cardiovascular dysfunction. Indeed, daytime hypertension has been induced in a canine model of OSA (12) as well as in rodent models of intermittent hypoxia (13). Thus, there are potential biologic mechanisms to explain why OSA might be an independent risk factor for cardiovascular disease.
Obstructive Sleep Apnea and Cardiovascular Disease: Circa 1970s-1980s
In the 1980’s, studies in Europe found cross-sectional associations between snoring as a surrogate for OSA and both hypertension and cardiovascular disease (14,15). Slightly earlier, evidence also was emerging of an association between obstructive sleep apnea and hypertension. These studies noted a greater than 50% prevalence of hypertension among patients with OSA (16,17) and conversely, a 20-30% prevalence of OSA was observed in patients with hypertension (18,19). Furthermore, several case control and cohort studies indicated that the risk of stroke or heart disease was 2.1 to 10.3 fold greater for those with snoring (20-23). Cardiac arrhythmias were frequently noted to occur in OSA patients as well (24).
Later during this time period, there were 3 retrospective studies analyzing the relationship between OSA and mortality. From the Henry Ford Sleep Disorders Center, the vital status of 385 male patients with OSA studied with polysomnography between 1978 and 1986 was determined. Mortality was significantly greater in those with an apnea index (not apnea hypopnea index [AHI]) greater than 20 events/hour. Uvulopalatopharyngoplasty did not attenuate the mortality rate, but better survival was reported in those who had a tracheostomy or were prescribed nasal continuous positive airway pressure (CPAP) (25). At the same time, the Stanford Sleep Disorders Center analyzed their experience in 198 patients who had either a tracheostomy (n=71) or were treated conservatively with a recommendation for weight loss (n=127). There were 14 deaths of which 8 were from stroke or myocardial infarction over a 5 year follow-up period. All occurred in the conservatively treated group (26). In contrast, another series from the University of Florida failed to find any mortality differences between 91 treated and untreated OSA patients and 35 patients with symptoms consistent with OSA, but negative findings on polysomnography over a 7-98 month follow-up (27).
Given the known acute effects of repetitive obstruction of the upper airway on systemic and pulmonary blood pressure, and heart rate, as well as the recurrent episodes of hypoxemia (11), linkages between these physiologic findings and the development of cardiovascular disease were proposed (28). One such pathophysiologic pathway highlighted important roles for hypoxemia and increased sympathetic activity, both of which are currently considered important mechanistic factors for the development of cardiovascular disease (28).
Obstructive Sleep Apnea And Cardiovascular Disease 2012: Epidemiology
Starting in the mid 1990’s, important observations were made that solidified linkages between OSA and cardiovascular disease. In the Wisconsin Sleep Cohort, the first large prospective population-based cohort study to use polysomnography to confirm the presence of OSA, Peppard et al. (29) demonstrated that OSA was an independent risk factor for the development of hypertension. Furthermore, the risk progressively increased with greater levels of OSA severity (OR: 1.42, 2.03, 2.89 [AHI: <5, 5-<15, >15 /hour vs. referent=0]) (29). These findings were subsequently confirmed in the Sleep Heart Health Study (OR: 1.13, 1.54, 2.119 [AHI: 5-<15, 15-<30, >30 /hour vs. referent=<5], although they were significantly attenuated when body mass index (BMI) was included in the analytic models (30). In addition, the development of hypertension was found to be associated with the presence of nocturnal hypoxemia. A more recent prospective study from a clinical cohort showed similar findings (31). Moreover, a number of studies have shown that blood pressure will decrease after treatment of OSA with continuous positive airway pressure (32). Although the magnitude of improvement in large clinical trials is only 2-3 mm Hg, such changes are large from an epidemiologic and public health perspective, and may be greater in individual patients and those with resistant hypertension (33). Given this evidence, the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure has concluded that OSA is an identifiable cause for the development of hypertension (34).
Evidence now indicates that OSA is an independent risk factor for the development of coronary heart disease (CHD). In a Spanish clinical cohort of only men, there was an increased risk of incident CHD over a 12 year follow-up period (35). Subsequently, this observation was confirmed in the Sleep Heart Health Study (36). In this latter study, however, increased risk (Hazard Ratio: 1.75 for AHI > 30 /hour vs. AHI < 5) was only observed in men less than 70 years of age. It is possible that the absence of an impact of OSA on risk of CHD in older men is related to a “healthy survivor” effect (37). Thus, whether OSA increases the likelihood of developing CHD in older men and women remains unclear.
In addition to findings linking OSA to greater risk of incident CHD, OSA appears to enhance the likelihood of new events in those with prevalent CHD. In a study of 407 consecutive patients with CHD, those with an oxygen desaturation index greater than 5 per hour had a 70% relative increase and a 10.7% absolute increase in the composite endpoint (cerebrovascular event, myocardial infarction or death) (38). In another study, 89 patients who had undergone percutaneous coronary intervention had polysomnography with OSA found in 51. In comparison to those who did not have OSA, 23.5% vs. 5.3% had a major adverse coronary event (cardiac death, reinfarction, and target vessel revascularization) in the ensuing follow-up period (mean=227 days) (39).
Sleep apnea, in particular central sleep apnea, frequently is observed in association with congestive heart failure (CHF). However, recent data from the Sleep Heart Health Study suggest that severe OSA is a risk factor for incident CHF in men (Hazard Ratio: 1.71 for AHI > 30 /hour vs. AHI < 5), but not women (36).
Recent observations indicate that stroke incidence also may be increased in those with OSA. In the Wisconsin Sleep Cohort over a 4 year follow-up, a significantly increased odds ratio for incident stroke after age and sex adjustment of 4.48 was observed in those with an AHI > 20 /hour (40). This was attenuated to 3.08 after controlling for body mass index. In the Sleep Heart Health Study, a significant increase in incident stroke risk also was found in those with an AHI > 20 /hour, but only in men (41).
As initially reported over 25 years ago (24), more recent observations have confirmed an association between OSA and cardiac arrhythmias. In the Sleep Heart Health Study, those with severe OSA were more likely to have both ventricular and atrial ectopy (42). Furthermore, those with severe OSA were 4.5 and 1.8 times more likely to have episodes of atrial fibrillation, and complex ventricular ectopy or non-sustained ventricular tachycardia.42 Additional analyses reveal that the relative risk of having an episodes of atrial fibrillation is 17 times greater after an apnea or hypopnea episode and there is 1 excess episode of paroxysmal atrial fibrillation or nonsustained ventricular tachycardia for every 1000 hours of sleep or 40000 respiratory disturbances (43). Obstructive sleep apnea with attendant hypoxemia also may be a risk factor for recurrence of atrial fibrillation after cardioversion (44).
Finally, there is now relatively conclusive evidence from 3 longitudinal cohort studies demonstrating that OSA contributes to excess mortality. In the Busselton Health Study of 380 individuals followed for a mean of 13.4 years, a respiratory disturbance index of greater than 15 /hour yielded a hazard ratio of 6.24 for excess mortality (45). Subsequently, in an 18 year follow-up of 1496 participants in the Wisconsin Sleep Cohort, the adjusted hazard ratio for excess all cause mortality related to severe OSA was 3.0 in comparison to no OSA (46). Furthermore, the hazard ratio related to cardiovascular disease mortality was 5.2 (46). More recently, the Sleep Heart Health Study reported a hazard ratio of 1.46 for all cause mortality over an 8.2 year average follow-up. Similar to the Wisconsin Sleep Cohort, it appeared that cardiovascular deaths accounted for much of the excess risk (47). In this latter study, indices of nocturnal hypoxemia also were associated with excess all cause mortality. Although sudden cardiac death in the general population usually occurs during the 6 am to 12 noon time frame, in those with OSA, it is shifted to night-time hours, 12 midnight to 6 am providing further evidence of the adverse impact of OSA on the heart (48).
Obstructive Sleep Apnea and Cardiovascular Disease: Mechanistic Observations
Since the initial observations of the physiologic events that might be operative in the pathogenesis of cardiovascular sequelae of OSA over 25 years ago, substantial progress has been made towards understanding how OSA is a risk factor for cardiovascular disease. These findings include OSA induced changes in cardiac structure and function, abnormalities in metabolic function, and increases in inflammation, coagulability and sympathetic nervous system activity. These latter issues then interact to enhance atherogenesis and cardiac dysfunction.
Obstructive sleep apnea is associated with increases in left ventricular mass. In the Sleep Heart Health Study, those with an AHI > 30 /hour in comparison to those with an AHI < 5 were more likely to have left ventricular hypertrophy on echocardiography, and estimates of left ventricular mass were higher (49). These associations were even stronger when indices of nocturnal hypoxemia were used instead of the AHI. This further highlights the potential role of hypoxemia in the pathogenesis of cardiovascular disease attributable to OSA. Given the presence of left ventricular hypertrophy related to OSA, it is not surprising that diastolic dysfunction is more common among individuals with OSA (50). However, use of CPAP may improve left ventricular function (50). This raises the possibility that early intervention to treat OSA may reduce cardiac morbidity and mortality.
A number of studies have demonstrated that OSA is associated with metabolic abnormalities. For example, in the Sleep Heart Health Study, levels of cholesterol and triglycerides increased as a function of increasing OSA severity (51). These findings may be related to OSA induced intermittent hypoxia (52). Furthermore, the prevalence of metabolic syndrome is higher among persons with OSA in comparison to those without OSA (53). This finding appears to be driven primarily by the higher frequency of hypertension in persons with OSA. Whether these findings are causally related to OSA remains to be determined. However, OSA might potentially increase the risk of CHD by promoting dyslipidemia.
The prevalence of OSA among persons with type 2 diabetes mellitus is high with one study observing that 86% of obese type 2 diabetics had an AHI >5 /hour, indicative of mild OSA (54). Therefore, it is not surprising that considerable evidence now implicates OSA as a determinant of glucose regulation. Both cross-sectional and prospective studies have demonstrated that OSA is a risk factor for glucose dysregulation and in some studies incident diabetes mellitus (55). Furthermore, some studies have demonstrated that treatment of OSA with CPAP results in improved glucose control (55). Data suggest that OSA induced hypoxemia may be a causative mechanism (56). The close association between OSA and type 2 diabetes mellitus raises the distinct possibility that a positive interaction exists to increase the risk of CHD in persons with both conditions.
It is generally accepted that obesity is a risk factor for the development of OSA (57). However, a few studies have reported that reverse causality may be present such that OSA promotes weight gain (58,59). Recent data from the Sleep Heart Health Study support this hypothesis. Over an approximate 5 year follow-up, weight gain was greater among those with an AHI > 15 /hour in comparison to those with an AHI < 5 /hour (60). Thus, promotion of weight gain may be another mechanism by which OSA increases cardiovascular risk.
Especially in those with severe OSA, recurring apneas and hypopneas result in repetitive episodes of hypoxia and reoxygenation. This produces oxidative stress leading to an increase in the flux of free radicals, induction of endothelin expression, suppression of nitric oxide generation, local vasoconstriction and changes in vascular permeability (61). All of these effects have the potential of enhancing the development of cardiovascular disease.
Substantial data now is available demonstrating that OSA is associated with release of a number of inflammatory mediators such as IL6, sIL6R, IL-8, TNFα, CRP and NF-Kappa β (62). In addition, there is evidence for elevated levels of pro-thrombotic factors such as PAI-1, P-selectin, fibrinogen and VEGF in persons with OSA (62). With the recent findings of the importance of inflammation and thrombosis in the pathogenesis of cardiovascular disease, these observations may be important causal mechanistic links that lead OSA to cardiovascular disease.
Using observations from 25 years ago in combination with current data, a plausible pathogenic pathway from OSA to cardiovascular can be summarized as follows (Figure 1). Recurrent episodes of apnea and hypopnea lead to intermittent hypoxia, increased sympathetic activity, hypercapnia, sleep fragmentation with arousals and large swings in intrathoracic pressure. These physiologic perturbations result in increased oxidative stress, release of inflammatory mediators, metabolic dysfunction, weight gain, hypercoagulability, glucose dysregulation and endothelial dysfunction All of these mechanisms can lead to the development of hypertension, diabetes mellitus, CHD, CHF, stroke and increased risk of death.
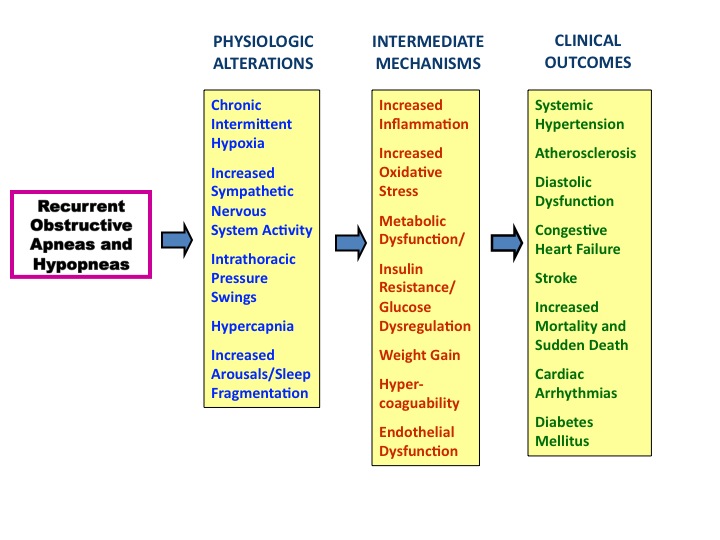
Figure 1. Proposed mechanisms leading from physiologic alterations occurring during obstructive sleep apnea and hypopnea to the development of cardiovascular disease.
Obstructive Sleep Apnea and Cardiovascular Disease: Knowledge Gaps
Although significant advances have been made in our understanding of the relationship between OSA and CVD in the past 25 years, there are a number of important areas which require further investigation. With respect to OSA and hypertension, it remains unclear whether treatment of OSA reduces the risk of developing hypertension. Most studies to date have used non-randomized cohorts. In the most recently published randomized controlled trial, CPAP treatment did not decrease the incidence of hypertension in nonsleepy subjects over a median 4 year follow-up (63). However, post-hoc analyses did suggest an effect in subjects who were compliant with CPAP for more than 4 hours per night (63). Furthermore, if CPAP or other treatments for OSA are beneficial in reducing CVD risk, are there subsets of the population for whom it is more advantageous?
As to the impact of OSA on CVD and stroke, there also have not been any published large scale clinical trials demonstrating an impact of treatment on changing the incidence of disease. Similar to hypertension, published studies are from non-randomized cohorts. However, there are several large clinical trials such as Randomized Intervention with CPAP in Coronary Artery Disease and Sleep Apnoea (RICCADSA), Sleep Apnea Cardiovascular Endpoints Study (SAVE), and Heart Biomarker Evaluation in Apnea Treatment study (HeartBEAT). In the RICCADSA study, 400 CAD participants will be randomized to one of 4 groups: 1) non-sleepy with OSA treated with CPAP, 2) non-sleepy with OSA and no CPAP treatment, 3) sleepy with OSA treated with CPAP, 4) CAD but no OSA. The participants will be followed for 3 years for CVD morbidity and mortality (64). In the multinational SAVE trial, participants with OSA at high risk for CVD will be randomized to CPAP or conventional medical therapy, and followed for 3-5 years (65). Because of the length of follow-up required and expense, and some would argue the ethical dilemmas in performing a long-term interventional trial, studies such as the recently completed HeartBEAT have attempted to assess intermediate outcomes. In the HeartBEAT trial, 270 subjects with CHD or at high risk for CHD were randomized to healthy lifestyle instruction, CPAP or nocturnal oxygen with a primary endpoint of 24 hour blood pressure (66). Whether findings from trials using intermediate endpoints will be predictive of an impact on “hard” endpoints such as incident myocardial infarction or stroke remains to be determined.
Conclusions
Substantial progress has been made in the past 25-30 years in our understanding of the relationship between OSA and CVD. Accumulating evidence implicates OSA as an independent risk factor for hypertension, CHD and stroke. However, the risk may not be the same for all segments of the population. A variety of mechanisms may be operative. Non-randomized trials suggest that treatment appears to mitigate the risk in some clinical populations, but it is unclear whether treatment is beneficial in patients without symptoms. Large scale randomized clinical trials are needed to clearly demonstrate that current treatment modalities for OSA can mitigate CVD risk and to delineate which populations will accrue the most benefit.
References
- Gastaut H, Tassinari CA, Duron B. Polygraphic study of diurnal and nocturnal (hypnic and respiratory) episodal manifestations of Pickwick syndrome. Rev Neurol (Paris) 1965; 112(6):568-579.
- Kryger MH. Sleep apnea. From the needles of Dionysius to continuous positive airway pressure. Arch Intern Med 1983; 143(12):2301-2303.
- Bickelmann AG, Burwell CS, Robin ED, Whaley RD. Extreme obesity associated with alveolar hypoventilation; a Pickwickian syndrome. Am J Med 1956; 21(5):811-818.
- White DP. Sleep apnea. Proc Am Thorac Soc 2006; 3(1):124-128.
- Podszus T, Greenberg H, Scharf SM. Influence of Sleep State and Sleep-Disordered Brathing on Cardiovascular Function. In: Sleep and Breathing. Saunders NA and Sullivan CE, eds. New York, NY: Marcel Dekker, 1994; 257-310
- Schaub CD, Schneider H, O'Donnell CP. Mechanisms of acute and chronic blood pressure elevationin animal models of obstructive sleep apnea. In: Sleep apnea: implications in cardiovascular and cerebrovascular disease. Bradley TD and Floras JS, eds. New York: Dekker, 2000; 554
- Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 1995; 96(4):1897-1904.
- Leung RS. Sleep-disordered breathing: autonomic mechanisms and arrhythmias. Prog Cardiovasc Dis 2009; 51(4):324-338.
- Parker JD, Brooks D, Kozar LF, et al. Acute and chronic effects of airway obstruction on canine left ventricular performance. Am J Respir Crit Care Med 1999; 160(6):1888-1896.
- Lorenzo-Filho G, Bradley TD. Cardiac function in sleep apnea. In: Sleep Apnea--Pathogeneis, Diagnosis, and Treatment. Pack AI, ed. New York, NY: Marcel Dekker, 2002; 377-410
- Schroeder JS, Motta J, Guilleminault C. Hemodynamic studies in sleep apnea. In: Sleep Apnea Syndromes. Guilleminault C and Dement WC, eds. New York: Alan R. Liss, 1978; 177-196
- Brooks D, Horner RL, Kozar LF, Render-Teixeira CL, Phillipson EA. Obstructive sleep apnea as a cause of systemic hypertension. Evidence from a canine model. J Clin Invest 1997; 99(1):106-109.
- Bosc LV, Resta T, Walker B, Kanagy NL. Mechanisms of intermittent hypoxia induced hypertension. J Cell Mol Med 2010; 14(1-2):3-17.
- Lugaresi E, Cirignotta F, Coccagna G, Piana C. Some epidemiological data on snoring and cardiocirculatory disturbances. Sleep 1980; 3(3-4):221-224.
- Koskenvuo M, Kaprio J, Partinen M, Langinvainio H, Sarna S, Heikkila K. Snoring as a risk factor for hypertension and angina pectoris. Lancet 1985; 1(8434):893-896.
- Guilleminault C, Tilkian A, Dement WC. The sleep apnea syndromes. Annu Rev Med 1976; 27:465-484.
- Kales A, Cadieux RJ, Bixler EO, et al. Severe obstructive sleep apnea--I: Onset, clinical course, and characteristics. J Chronic Dis 1985; 38(5):419-425.
- Lavie P, Ben-Yosef R, Rubin AE. Prevalence of sleep apnea syndrome among patients with essential hypertension. Am Heart J 1984; 108(2):373-376.
- Fletcher EC, DeBehnke RD, Lovoi MS, Gorin AB. Undiagnosed sleep apnea in patients with essential hypertension. Ann Intern Med 1985; 103(2):190-195.
- Spriggs DA, French JM, Murdy JM, Bates D, James OF. Historical risk factors for stroke: a case control study. Age Ageing 1990; 19(5):280-287.
- Koskenvuo M, Kaprio J, Telakivi T, Partinen M, Heikkila K, Sarna S. Snoring as a risk factor for ischaemic heart disease and stroke in men. Br Med J (Clin Res Ed) 1987; 294(6563):16-19.
- Palomäki H, Partinen M, Juvela S, Kaste M. Snoring as a risk factor for sleep-related brain infarction. Stroke 1989; 20(10):1311-1315.
- Partinen M, Palomaki H. Snoring and cerebral infarction. Lancet 1985; 2(8468):1325-1326.
- Boudoulas H, Schmidt HS, Clark RW, Geleris P, Schaal SF, Lewis RP. Anthropometric characteristics, cardiac abnormalities and adrenergic activity in patients with primary disorders of sleep. J Med 1983; 14(3):223-238.
- He J, Kryger MH, Zorick FJ, Conway W, Roth T. Mortality and apnea index in obstructive sleep apnea. Experience in 385 male patients. Chest 1988; 94(1):9-14.
- Partinen M, Jamieson A, Guilleminault C. Long-term outcome for obstructive sleep apnea syndrome patients. Mortality. Chest 1988; 94(6):1200-1204.
- Gonzalez-Rothi RJ, Foresman GE, Block AJ. Do patients with sleep apnea die in their sleep? Chest 1988; 94(3):531-538.
- Tilkian AG, Guilleminault C, Schroeder JS, Lehrman KL, Simmons FB, Dement WC. Hemodynamics in sleep-induced apnea. Studies during wakefulness and sleep. Ann Intern Med 1976; 85(6):714-719.
- Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 2000; 342(19):1378-84.
- O'Connor GT, Caffo B, Newman AB, et al. Prospective study of sleep-disordered breathing and hypertension: the Sleep Heart Health Study. Am J Respir Crit Care Med 2009; 179(12):1159-1164.
- Marin JM, Agusti A, Villar I, et al. Association between treated and untreated obstructive sleep apnea and risk of hypertension. JAMA 2012; 307(20):2169-2176.
- Bazzano LA, Khan Z, Reynolds K, He J. Effect of nocturnal nasal continuous positive airway pressure on blood pressure in obstructive sleep apnea. Hypertension 2007; 50(2):417-423.
- Lozano L, Tovar JL, Sampol G, et al. Continuous positive airway pressure treatment in sleep apnea patients with resistant hypertension: a randomized, controlled trial. J Hypertens 2010; 28(10):2161-2168.
- Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42(6):1206-1252.
- Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 2005; 365(9464):1046-1053.
- Gottlieb DJ, Yenokyan G, Newman AB, et al. Prospective study of obstructive sleep apnea and incident coronary heart disease and heart failure: the sleep heart health study. Circulation 2010; 122(4):352-360.
- Murphy TE, Han L, Allore HG, Peduzzi PN, Gill TM, Lin H. Treatment of death in the analysis of longitudinal studies of gerontological outcomes. J Gerontol A Biol Sci Med Sci 2011; 66(1):109-114.
- Mooe T, Franklin KA, Holmstrom K, Rabben T, Wiklund U. Sleep-disordered breathing and coronary artery disease: long-term prognosis. Am J Respir Crit Care Med 2001; 164(10 Pt 1):1910-1913.
- Yumino D, Tsurumi Y, Takagi A, Suzuki K, Kasanuki H. Impact of obstructive sleep apnea on clinical and angiographic outcomes following percutaneous coronary intervention in patients with acute coronary syndrome. Am J Cardiol 2007; 99(1):26-30.
- Arzt M, Young T, Finn L, Skatrud JB, Bradley TD. Association of sleep-disordered breathing and the occurrence of stroke. Am J Respir Crit Care Med 2005; 172(11):1447-1451.
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive Sleep Apnea Hypopnea and Incident Stroke: The Sleep Heart Health Study. Am J Respir Crit Care Med 2010;
- Mehra R, Benjamin EJ, Shahar E, et al. Association of nocturnal arrhythmias with sleep-disordered breathing: The Sleep Heart Health Study. Am J Respir Crit Care Med 2006; 173(8):910-6.
- Monahan K, Storfer-Isser A, Mehra R, et al. Triggering of nocturnal arrhythmias by sleep-disordered breathing events. J Am Coll Cardiol 2009; 54(19):1797-1804.
- Kanagala R, Murali NS, Friedman PA, et al. Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation 2003; 107(20):2589-2594.
- Marshall NS, Wong KK, Liu PY, Cullen SR, Knuiman MW, Grunstein RR. Sleep apnea as an independent risk factor for all-cause mortality: the Busselton Health Study. Sleep 2008; 31(8):1079-1085.
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep 2008; 31(8):1071-1078.
- Punjabi NM, Caffo BS, Goodwin JL, et al. Sleep-disordered breathing and mortality: a prospective cohort study. PLoS Med 2009; 6(8):e1000132.
- Gami AS, Howard DE, Olson EJ, Somers VK. Day-night pattern of sudden death in obstructive sleep apnea. N Engl J Med 2005; 352(12):1206-1214.
- Chami HA, Devereux RB, Gottdiener JS, et al. Left ventricular morphology and systolic function in sleep-disordered breathing: the Sleep Heart Health Study. Circulation 2008; 117(20):2599-2607.
- Arias MA, Garcia-Rio F, Alonso-Fernandez A, Mediano O, Martinez I, Villamor J. Obstructive sleep apnea syndrome affects left ventricular diastolic function: effects of nasal continuous positive airway pressure in men. Circulation 2005; 112(3):375-383.
- Newman AB, Nieto FJ, Guidry U, et al. Relation of sleep-disordered breathing to cardiovascular disease risk factors: the Sleep Heart Health Study. Am J Epidemiol 2001; 154(1):50-59.
- Adedayo AM, Olafiranye O, Smith D, et al. Obstructive sleep apnea and dyslipidemia: evidence and underlying mechanism. Sleep Breath 2012;
- Parish JM, Adam T, Facchiano L. Relationship of metabolic syndrome and obstructive sleep apnea. J Clin Sleep Med 2007; 3(5):467-472.
- Foster GD, Sanders MH, Millman R, et al. Obstructive sleep apnea among obese patients with type 2 diabetes. Diabetes Care 2009; 32(6):1017-1019.
- Clarenbach CF, West SD, Kohler M. Is obstructive sleep apnea a risk factor for diabetes? Discov Med 2011; 12(62):17-24.
- Punjabi NM, Shahar E, Redline S, et al. Sleep-disordered breathing, glucose intolerance, and insulin resistance: the Sleep Heart Health Study. Am J Epidemiol 2004; 160(6):521-530.
- Young T, Peppard PE, Taheri S. Excess weight and sleep-disordered breathing. J Appl Physiol 2005; 99(4):1592-1599.
- Traviss KA, Barr SI, Fleming JA, Ryan CF. Lifestyle-related weight gain in obese men with newly diagnosed obstructive sleep apnea. J Am Diet Assoc 2002; 102(5):703-706.
- Phillips BG, Hisel TM, Kato M, et al. Recent weight gain in patients with newly diagnosed obstructive sleep apnea. J Hypertens 1999; 17(9):1297-1300.
- Brown MA, Goodwin JL, Silva GE, et al. The Impact of Sleep-Disordered Breathing on Body Mass Index (BMI): The Sleep Heart Health Study (SHHS). Southwest J Pulm Crit Care 2011; 3:159-168.
- Prabhakar NR. Oxygen sensing during intermittent hypoxia: cellular and molecular mechanisms. J Appl Physiol 2001; 90(5):1986-1994.
- Kent BD, Ryan S, McNicholas WT. Obstructive sleep apnea and inflammation: relationship to cardiovascular co-morbidity. Respir Physiol Neurobiol 2011; 178(3):475-481.
- Barbe F, Duran-Cantolla J, Sanchez-de-la-Torre M, et al. Effect of continuous positive airway pressure on the incidence of hypertension and cardiovascular events in nonsleepy patients with obstructive sleep apnea: a randomized controlled trial. JAMA 2012; 307(20):2161-2168.
- Peker Y, Glantz H, Thunstrom E, Kallryd A, Herlitz J, Ejdeback J. Rationale and design of the Randomized Intervention with CPAP in Coronary Artery Disease and Sleep Apnoea--RICCADSA trial. Scand Cardiovasc J 2009; 43(1):24-31.
- Anonymous . Continuous Positive Airway Pressure Treatment of Obstructive Sleep Apnea to Prevent Cardiovascular Disease (SAVE). Last Updated: 2011. Accessed: October 1, 2012. http://www.clinicaltrials.gov/ct2/show/NCT00738179.
- Anonymous . Heart Biomarker Evaluation in Apnea Treatment (HeartBEAT). Last Updated: 2011. Accessed: October 1, 2012. http://www.clinicaltrials.gov/ct2/show/NCT01086800.
Contact Information:
Stuart F. Quan, M.D.
Division of Sleep Medicine
Harvard Medical School
401 Park Dr., 2nd Floor East
Boston, MA 02215
Voice: 617-998-8842
Fax: 617-998-8823
Email: Stuart_Quan@hms.harvard.edu
Conflicts of Interest: The author does not have any conflict of interests pertinent to the subject matter of this review.
Reference as: Quan SF. Obstructive sleep apnea and cardiovascular disease: back and forward in time over the last 25 years. Southwest J Pulm Crit Care 2012;5:206-17. PDF
 Post a Comment
Post a Comment
Reader Comments